臨床成績
「注意事項等情報」等は
電子添文をご参照ください。
海外第 II 相試験(UX007-CL201試験)(海外データ)1-4
UX007-CL201試験は、LC-FOAD患者†におけるドジョルビの有効性及び安全性を評価した海外第II相試験です。
ドジョルビ投与前後各78週間での主要臨床イベント(MCE)の年換算発現率及び年換算発現日数の変化などを主な有効性評価項目として検討しています。
- 極長鎖アシルCoA脱水素酵素(VLCAD)欠損症、長鎖3-ヒドロキシアシルCoA脱水素酵素(LCHAD欠損症)、カルニチンパルミトイルトランスフェラーゼ2(CPT2)欠損症及び三頭酵素(TFP)欠損症患者
MCE:major clinical events
MCEの年換算発現率
ドジョルビ投与前(78週間)及び投与開始後(78週間)におけるMCEの年換算発現率(主要解析対象集団)〈主な有効性評価項目〉
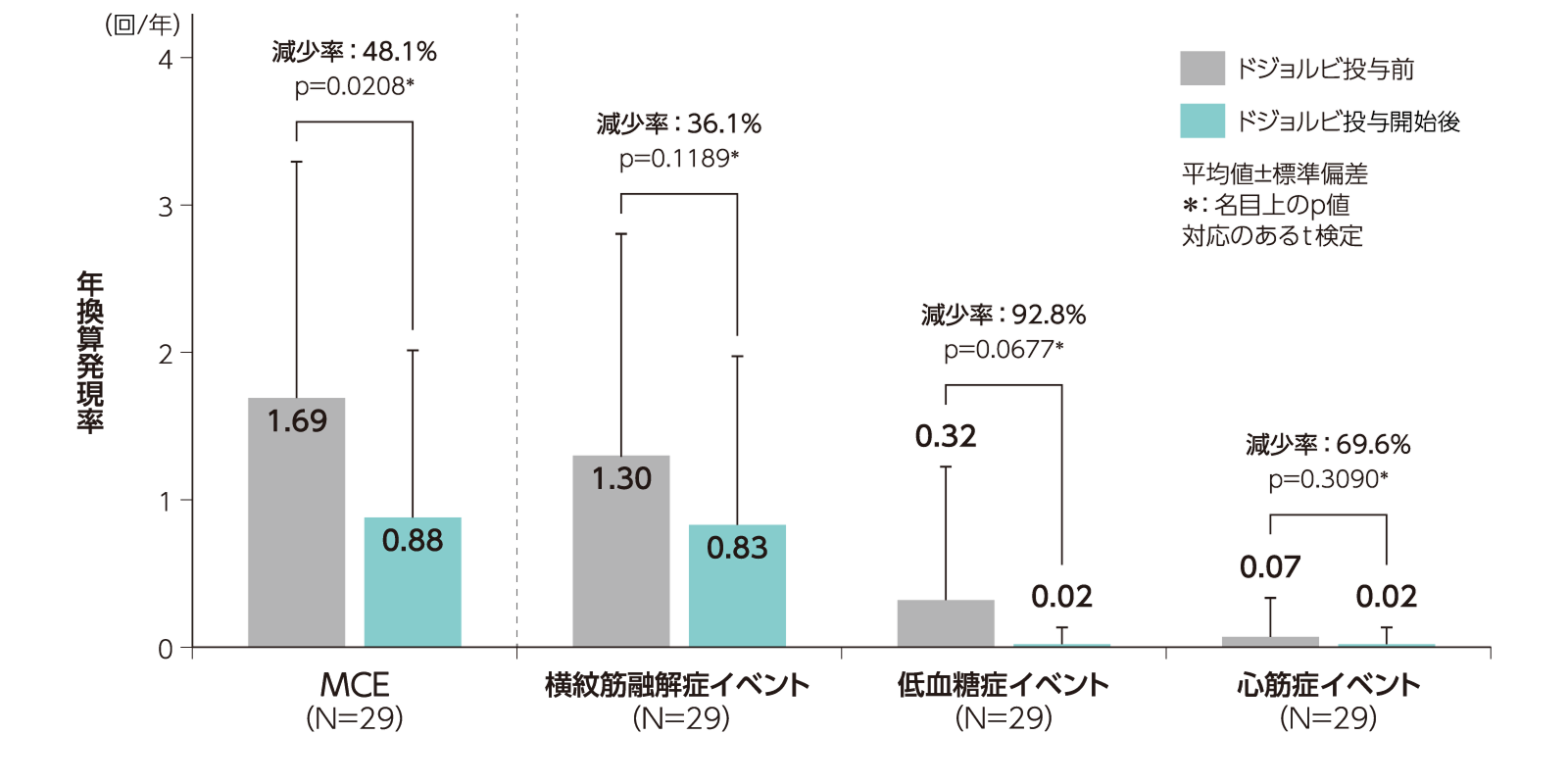
MCEaの年換算発現率(平均値)は、ドジョルビ投与前が1.69回/年、ドジョルビ投与開始後が0.88回/年bで、減少率は48.1%であった(p=0.0208、対応のあるt検定、名目上のp値)。
横紋筋融解症イベント、低血糖症イベント、心筋症イベントの年換算発現率(平均値)及び減少率は上図の通りであった。
MCEの年換算発現日数
ドジョルビ投与前(78週間)及び投与開始後(78週間)における各MCEの年換算発現日数(主要解析対象集団)〈主な有効性評価項目〉
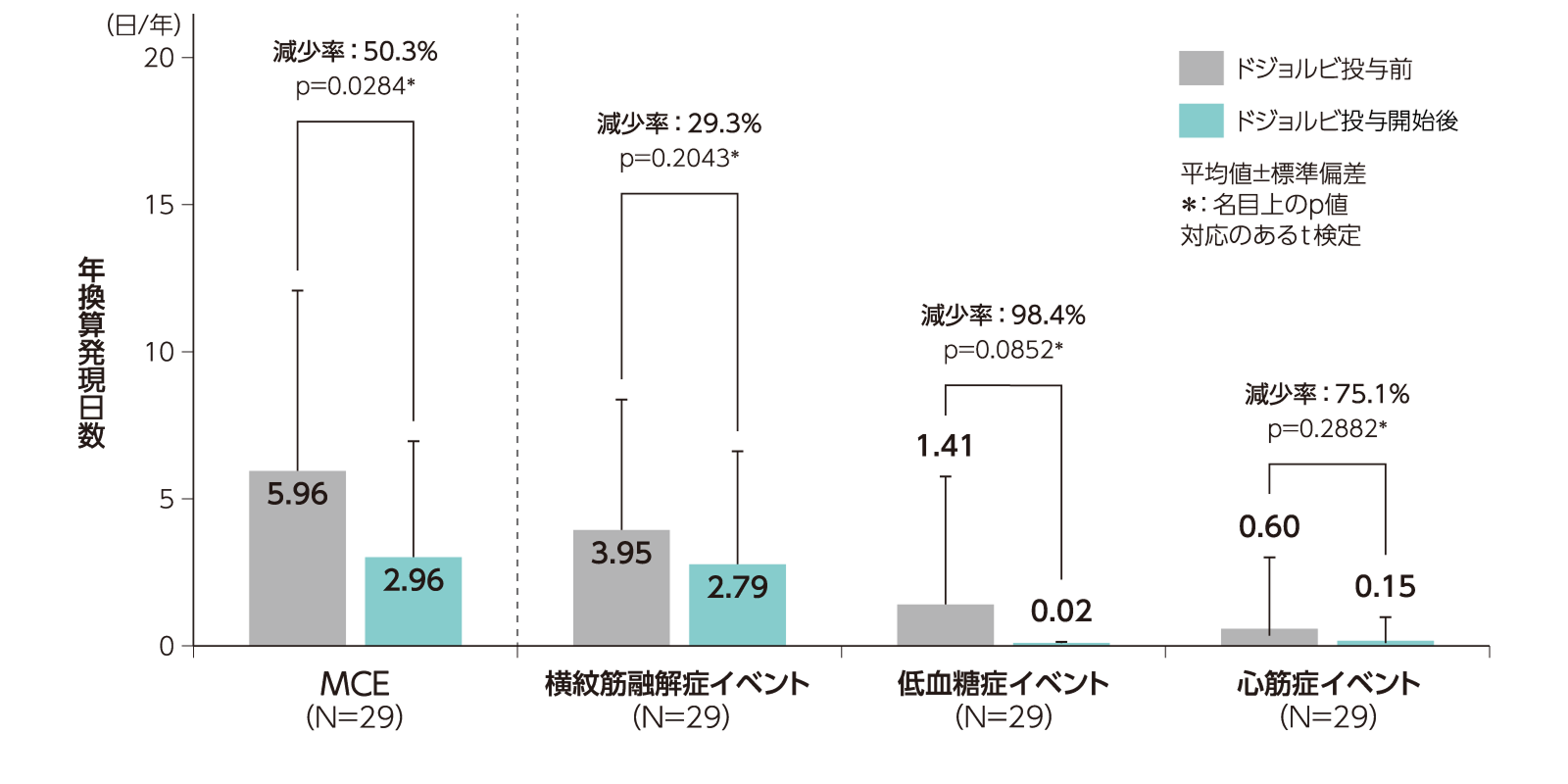
MCEaの年換算発現日数(平均値)は、ドジョルビ投与前が5.96日/年、ドジョルビ投与開始後が2.96日/年bで、減少率は50.3%であった(p=0.0284、対応のあるt検定、名目上のp値)。
横紋筋融解症イベント、低血糖症イベント、心筋症イベントの年換算発現日数(平均値)及び減少率は上図の通りであった。
入院に至ったMCEの年換算発現率
ドジョルビ投与前(78週間)及び投与開始後(78週間)における入院に至ったMCEの年換算発現率(主要解析対象集団)
〈主な有効性評価項目、副次解析a〉
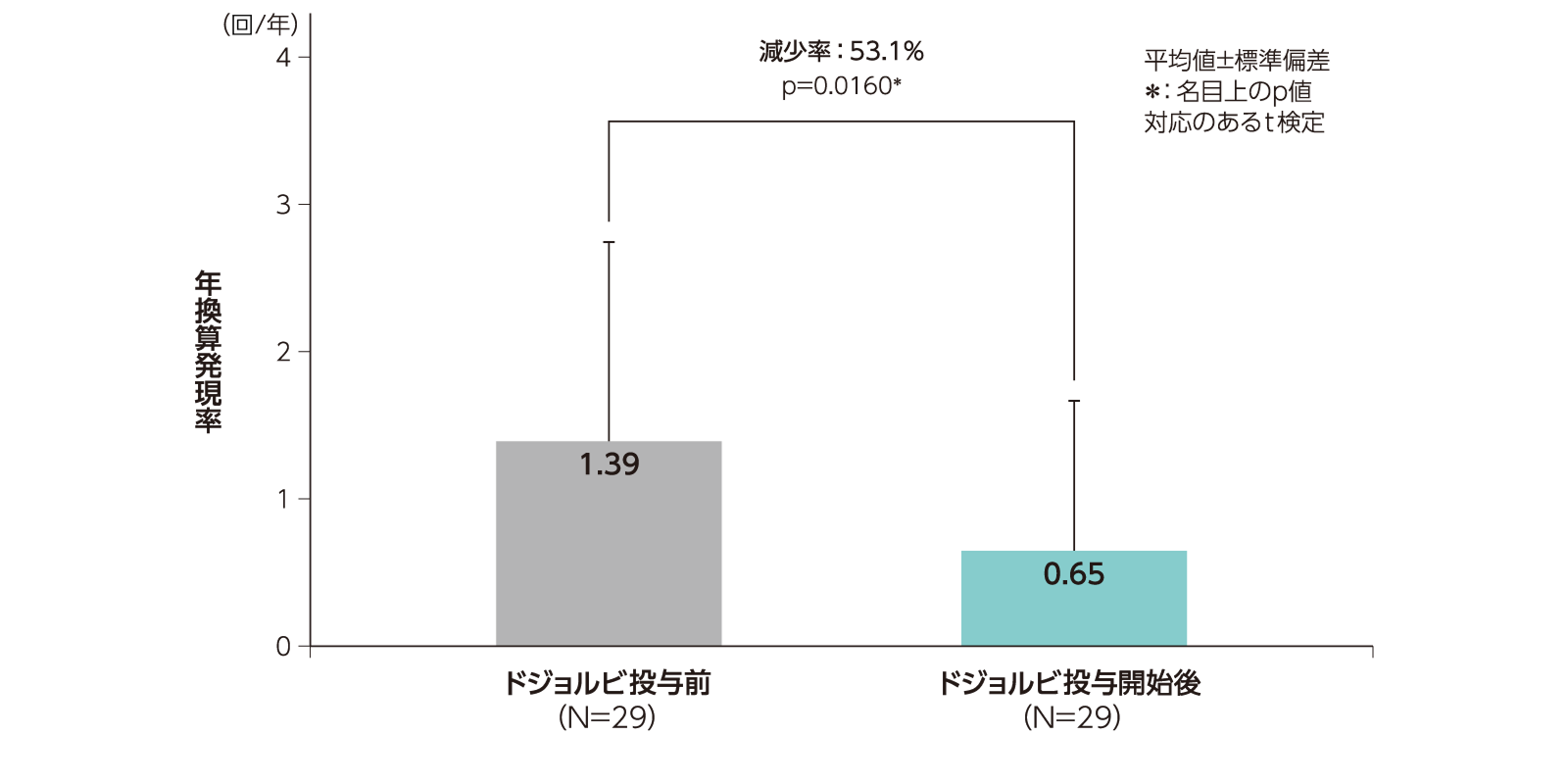
入院に至ったMCEbの年換算発現率(平均値)は、ドジョルビ投与前が1.39回/年、ドジョルビ投与開始後が0.65回/年で、減少率は53.1%であった(p=0.0160、対応のあるt検定、名目上のp値)。
入院に至ったMCEの年換算発現日数
ドジョルビ投与前(78週間)及び投与開始後(78週間)における入院に至ったMCEの年換算発現日数(主要解析対象集団)
〈主な有効性評価項目、副次解析a〉
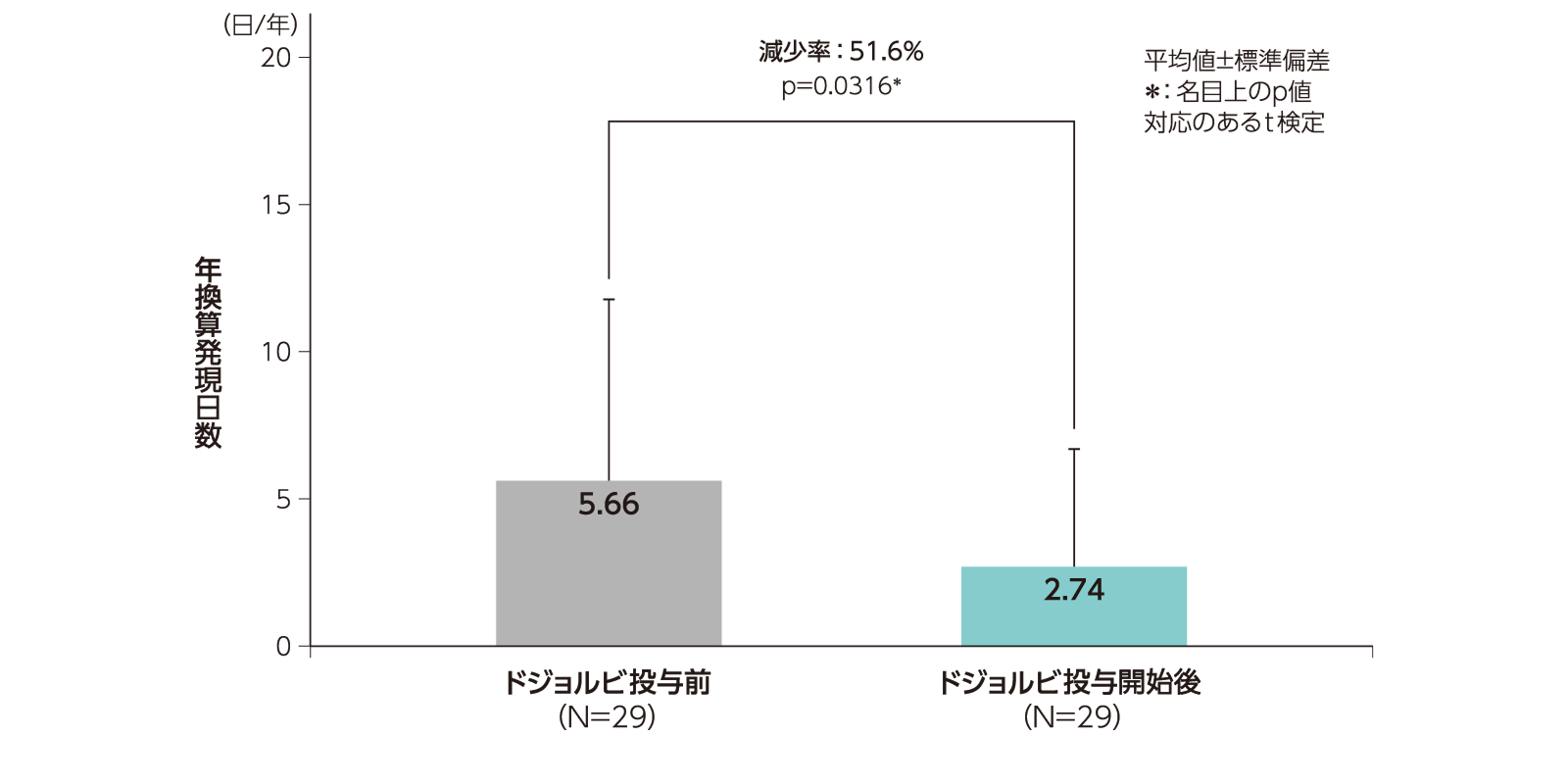
入院に至ったMCEbの年換算発現日数(平均値)は、ドジョルビ投与前が5.66日/年、ドジョルビ投与開始後が2.74日/年で、減少率は51.6%であった(p=0.0316、対応のあるt検定、名目上のp値)。
副作用
副作用(安全性解析対象集団)
| 発現例数(%) | |
|---|---|
| ドジョルビ群
(N=29) |
|
| 副作用発現例 | 19(65.5) |
| 下痢 | 12(41.4) |
| 腹痛 | 6(20.7) |
| 上腹部痛 | 3(10.3) |
| ざ瘡 | 3(10.3) |
| 消化器痛 | 3(10.3) |
| 嘔吐 | 3(10.3) |
| 腹部膨満 | 2(6.9) |
| 放屁 | 2(6.9) |
| 胃腸炎 | 2(6.9) |
| 悪心 | 2(6.9) |
| 血中トリグリセリド増加 | 1(3.4) |
| 気管支反応性亢進 | 1(3.4) |
| 便秘 | 1(3.4) |
| 食欲減退 | 1(3.4) |
| ウイルス性胃腸炎 | 1(3.4) |
| 胃食道逆流性疾患 | 1(3.4) |
| 左室肥大 | 1(3.4) |
| 筋痙縮 | 1(3.4) |
| 横紋筋融解症 | 1(3.4) |
| 体重増加 | 1(3.4) |
副作用aが1件以上発現した患者は、29例中19例(65.5%)であった(安全性解析対象集団)。主な副作用(発現頻度5%以上)は、下痢が12例(41.4%)、腹痛が6例(20.7%)、上腹部痛、ざ創、消化器痛及び嘔吐が各3例(10.3%)、腹部膨満、放屁、胃腸炎及び悪心が各2例(6.9%)であった。重篤な副作用が発現した患者は1例であり、胃腸炎が認められた。
副作用により治験薬の投与中止に至った患者は2例で、下痢及び嘔吐が各1例であった。死亡例はなかった。
目的
LC-FAOD(CPT2欠損症、VLCAD欠損症、LCHAD欠損症及びTFP欠損症)患者におけるドジョルビの有効性及び安全性を評価する。
主要目的:LC-FAODに関連する急性臨床病態生理に対するドジョルビの影響を評価する(24週間投与後)。
試験デザイン
後ろ向き及び前向き、介入、非盲検、単群、被験者自己対照試験
対象
現行の管理法aにもかかわらず、重篤な臨床症状を示す生後6ヵ月以上のLC-FAOD(CPT2欠損症、VLCAD欠損症、LCHAD欠損症及びTFP欠損症)患者29例
〈主な選択基準〉
- CPT2欠損症、VLCAD欠損症、LCHAD欠損症、TFP欠損症のいずれかと確定診断された者
- 生後6ヵ月以上の者
-
以下のいずれかの重大な臨床症状で裏付けられる重度LC-FAODである者
- MCEを伴う慢性的なクレアチンキナーゼ(CK)値の上昇
- 筋機能不全の報告を伴う一過性のCK値上昇
- 症状を伴わないCK高値
- 頻繁に発現する重度の主要医学的有害事象
- 低血糖症に対する重度な感受性
- 機能的心筋症の所見(過去90日以内の心エコー検査で駆出率の低下が確認され、継続的な医学的管理が必要)
〈主な除外基準〉
CPT1欠損症、CACT欠損症、中鎖アシルCoA脱水素酵素(MCAD)欠損症、短鎖又は中鎖FAOD、ケトン体代謝異常症、プロピオン酸血症又はメチルマロン酸血症と診断された者
方法
4週間の導入期間中、患者は現行の管理を継続した。導入期間終了後、ベースライン時に偶数鎖MCT(以降MCT)の使用を中止し、他の食事制限を維持しつつ、ドジョルビの投与を開始した。
ドジョルビの投与量は、患者の1日あたりのカロリー摂取量(DCI)aの25~35%を目標とし、1日4回(朝食、昼食、夕食、就寝前)に分け、食物又は飲料(乳児用粉ミルクを含む)に混合して経口又は経管投与した。
開始用量はDCIの約10%とし、目標投与量に至るまで忍容性を確認しながら漸増した。また、ベースライン時にMCTを使用していた患者では、MCTの量と同量からの開始を可能とした。ドジョルビへの忍容性が認められない場合は、1日あたりの投与回数を増加又は経管投与時の投与時間を延長し、それでも忍容性が認められない場合には減量が可能とされ、その後は最大耐用量を維持した。本試験では、以下の増量及び減量に係る規定が推奨されていた。また、各被験者の状況に応じて柔軟な対応も認められていた。
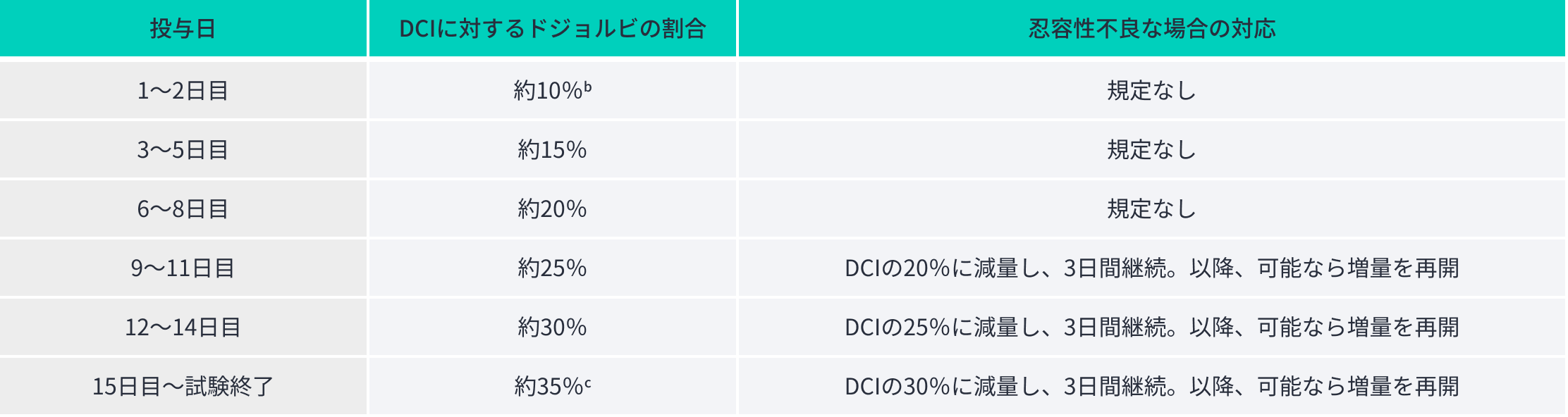
24週間(投与期間)投与し、その後、更に54週間の継続期間において投与を継続した(合計78週間)。
本試験では、ドジョルビ投与前後78週間のMCEd(定義については脚注d参照)発現について、患者内比較を行った。そのため、各患者が自己対照となるよう、ドジョルビ投与前期間(患者が従来のMCTを含む標準療法を受けていた期間)のデータを後ろ向きに収集したe。
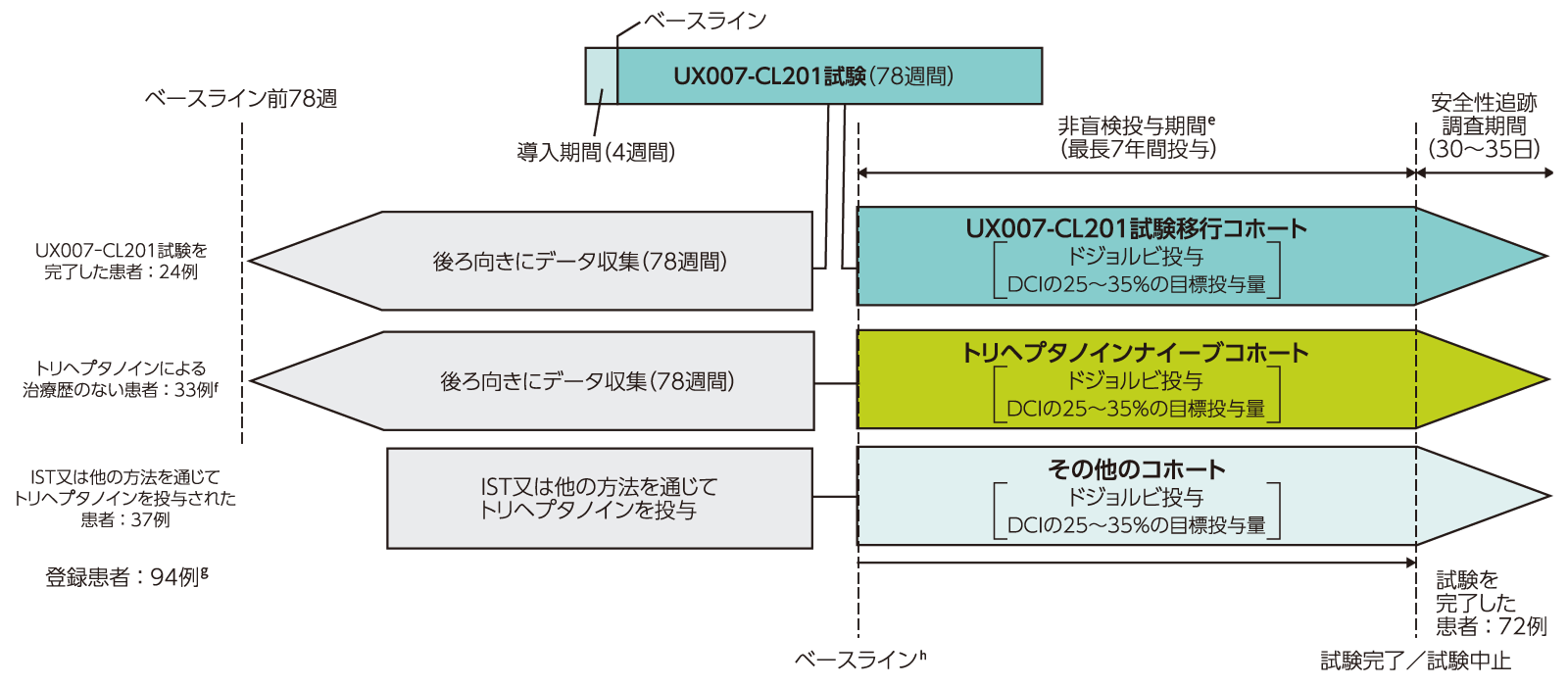
UX007-CL201試験のデザイン
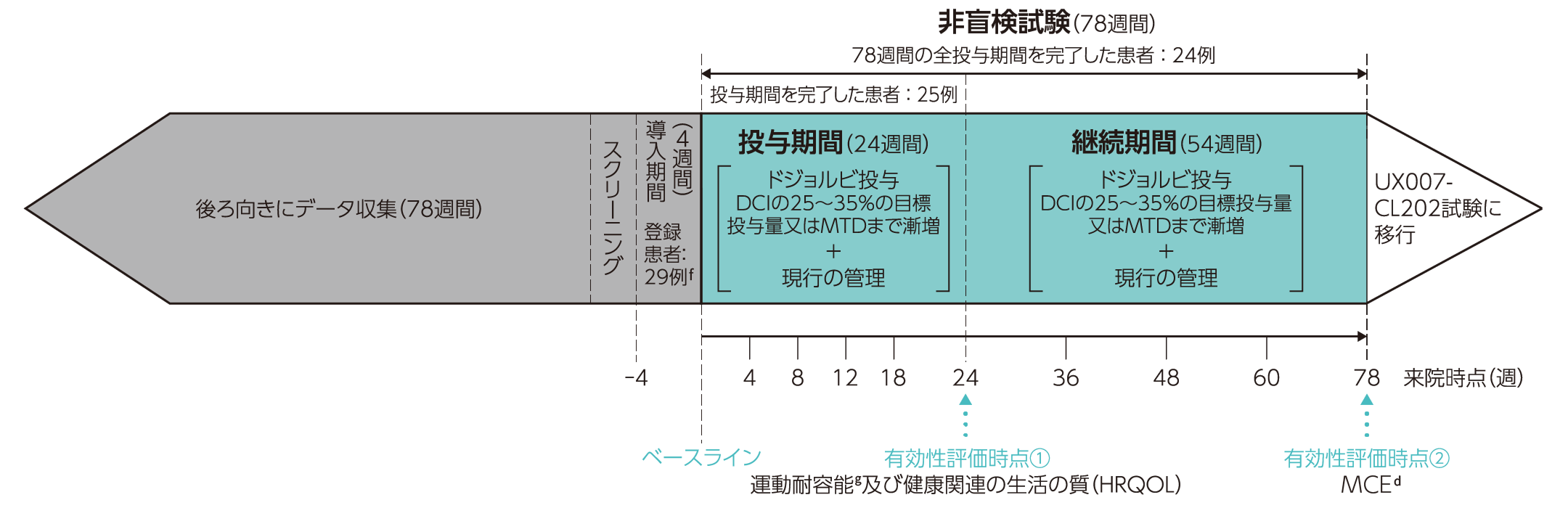
- 患者及び/又は介護者は、導入期間、ベースライン時及びドジョルビ投与後12、24、48、78週時の各来院前の3日間、食事日誌を記入した。食事日誌に基づき、1日あたりの平均カロリー摂取量を算出した。
- スクリーニング期間及び導入期間にMCTを摂取し、忍容性がある被験者では、ドジョルビをDCIの10%ではなく、MCTと同量から開始することを検討してよい。MCTと同量から開始する場合、ドジョルビをDCIの35%又は最大耐用量に達するまで、2日毎にDCIの約5%ずつ漸増する。
- 試験期間中、減量と再増量を行っても忍容性がなく、DCIの35%まで増量できない場合は、治験責任医師の判断による各被験者の最大耐用量を投与する。
- 横紋筋融解症、低血糖症又は心筋症のいずれかの事象の発現により、入院、緊急治療室若しくは救急科の受診又は緊急処置(自宅又は医療機関でのLC-FAODに対する予定外の治療薬の投与)に至った場合。
- 被験者自己対照として、ドジョルビ投与前78週間(18ヵ月間)又は生後18ヵ月未満の患者の場合は出生からドジョルビ投与開始までの入手可能なすべての診療記録を収集した。
- このうち、5例が試験を中止した(同意撤回4例、有害事象1例)。
- 運動耐容能評価のうち、12分間歩行テスト(12MWT)は評価スケジュールを考慮し、18週時の測定値を24週時の解析の主要時点の測定値とみなした。
評価項目
有効性について、主要評価項目は設定されず、主な有効性評価項目は以下の通りであった。
〈主な有効性評価項目〉
- ドジョルビ投与前(78週間)及びドジョルビ投与開始後(78週間)におけるMCEaの年換算発現率b及び年換算発現日数c
- MCE、横紋筋融解症イベント、低血糖症イベント、心筋症イベント、入院に至ったMCE
- 18週時における12分間歩行テスト(12MWT)の歩行距離のベースラインからの変化d
- 24週時におけるサイクルエルゴメーター検査値のベースラインからの変化d
- 仕事量及び持続時間
- 24週時におけるShort Form-12(SF-12)v2質問票のベースラインからの変化e(成人)
- 24週時におけるShort Form-10(SF-10)質問票のベースラインからの変化e(小児)
〈安全性評価項目〉
- 副作用 等
- 横紋筋融解症、低血糖症又は心筋症のいずれかの事象の発現により、入院、緊急治療室若しくは救急科の受診又は緊急処置(自宅又は医療機関でのLC-FAODに対する予定外の治療薬の投与)に至った場合。
- 年換算発現率=MCEの総数/[データ収集期間(日)÷365.25]。
- 年換算発現日数=MCEの総日数/[データ収集期間(日)÷365.25]。MCEに起因する総日数を、全患者を対象にデータ収集期間を1年間として平均化した期間と定義した。
- 検査の実施に信頼性があり安全に完了することができた6歳以上の患者を対象とした。
- 18歳以上の成人患者に対してSF-12v2質問票、5~17歳の小児患者の両親に対してSF-10質問票を用いて評価した。
解析計画
〈解析対象集団〉
| 定義 | 例数 | |
|---|---|---|
| 最大の解析対象集団(FAS) | 本試験に登録されたすべての患者 | 29 |
| 主要解析対象集団 | FASのうち、4週間の導入期間を完了し、かつドジョルビを少なくとも1回投与された患者 | 29 |
| 安全性解析対象集団 | ドジョルビを少なくとも1回投与されたすべての患者 | 29 |
〈主な有効性評価項目〉
有効性データの解析には、概して主要解析対象集団を用いた。
ドジョルビ投与開始後における主要臨床イベントの年換算発現率及び年換算発現日数は、対応のあるt検定を用いて、ドジョルビ投与前と比較した。
12MWTの歩行距離、サイクルエルゴメーター検査、SF-12v2質問票及びSF-10質問票を含むベースラインからの変化量は、時間をカテゴリー変数とし、ベースライン時の測定値で調整した一般化推定方程式(GEE)モデルを用いて解析した。GEEモデルに用いた共分散構造は複合対称とした。24週時の解析では、24週までの測定値をモデルに含めた。12MWTについては評価スケジュールを考慮し、18週時の測定値を24週時の解析の主要時点の測定値とみなした。GEEモデルを用いた解析の測定数が不十分な場合は、記述的な要約とし、対応のあるt検定又はその他のノンパラメトリックな方法等単一時点での測定値を用いた解析を行った。
検定は有意水準を両側5%とし、多重性の調整は実施しなかった。
〈安全性評価項目〉
安全性データの解析には、安全性解析対象集団を用いた。安全性データは記述的に要約した。副作用名はMedDRA version 17.1でコーディングした。
患者背景
患者背景(主要解析対象集団)
| ドジョルビ群(N=29) | ||
|---|---|---|
| 年齢a [歳] |
平均値±標準偏差 中央値 (最小値、最大値) |
12.1±13.2 5.3 (0.9,58.8) |
| 年齢層 [n(%)] |
6ヵ月以上6歳未満 6歳以上18歳未満 18歳以上 |
15(51.7) 8(27.6) 6(20.7) |
| 性別 [n(%)] |
男性 女性 |
17(58.6) 12(41.4) |
| 人種 [n(%)] |
白人 黒人又は アフリカ系アメリカ人 アジア人 その他 |
25(86.2) 1(3.4) 2(6.9) 1(3.4) |
| LC-FAOD サブタイプ [n(%)] |
CPT2欠損症 VLCAD欠損症 LCHAD欠損症 TFP欠損症 |
4(13.8) 12(41.4) 10(34.5) 3(10.3) |
| MCT使用歴 [n(%)] |
あり なし |
27(93.1) 2(6.9) |
参考文献
- 社内資料:UX007-CL201試験/海外第Ⅱ相試験(CTD2.7.3.2.1、2.7.4.2.1、2.7.6.2.2、5.3.5.2)[承認時評価資料]
- Vockley J, et al.: Mol Genet Metab. 2017; 120(4): 370-377.(利益相反:Ultragenyx Pharmaceutical, Incの支援により実施)
- Vockley J, et al.: J Inherit Metab Dis. 2019; 42(1): 169-177.(利益相反:Ultragenyx Pharmaceutical, Incの支援により実施)
- Vockley J, et al.: Clin Nutr ESPEN. 2021; 41: 293-298.(利益相反:Ultragenyx Pharmaceutical, Incの支援により実施)
海外第 II 相試験(UX007-CL202試験)(海外データ)1-3
UX007-CL202試験は、LC-FOADのすべてのサブタイプ患者†を対象としてドジョルビの有効性及び安全性を評価した海外第Ⅱ相試験です。
UX007-CL201試験から移行した患者(UX007-CL201試験移行コホート)およびトリヘプタノインの治療歴がない患者(トリヘプタノインナイーブコホート)にドジョルビを最長7年間投与した場合のMCEの年換算発現率を主要評価項目として検討しています。
- CPT1若しくはCTP2欠損症、VLCAD欠損症、LCHAD欠損症、TFP欠損症又はCACT欠損症のいずれか
MCEの年換算発現率および発現日数
UX007-CL201試験移行コホートにおけるMCEの年換算発現率及び年換算発現日数(FAS)〈主要評価項目及び副次評価項目〉
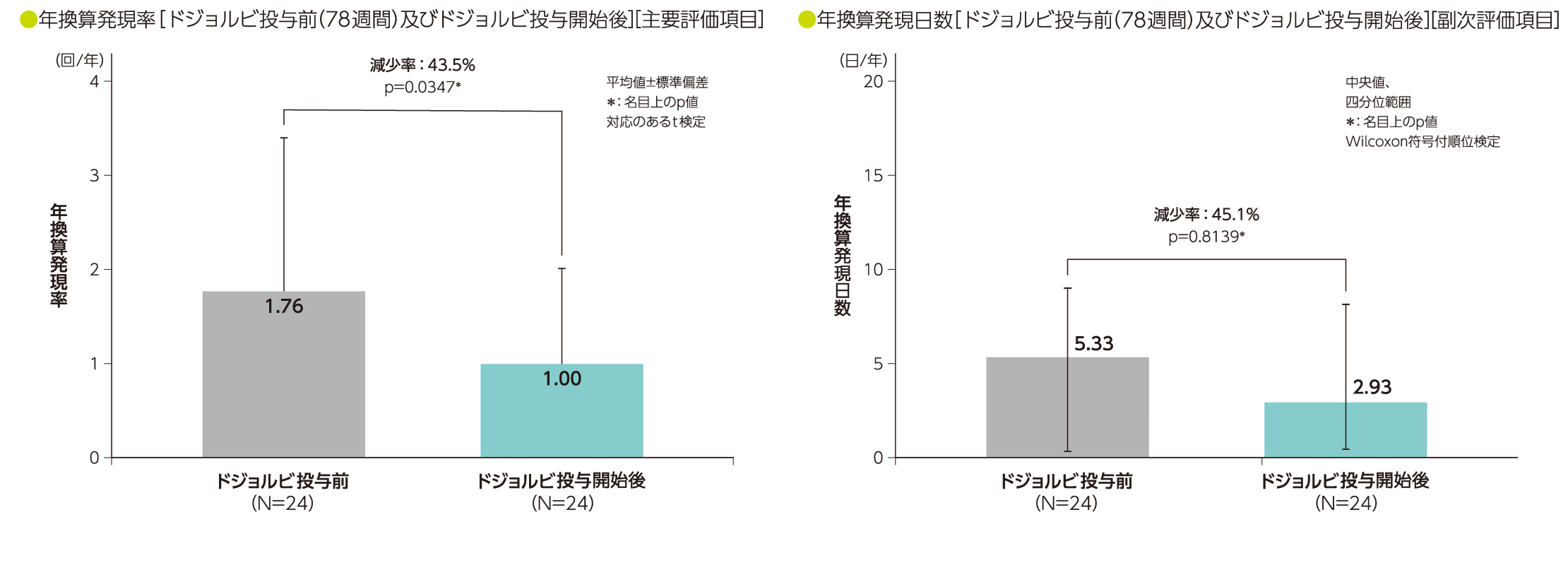
UX007-CL201試験移行コホートにおいて、主要評価項目であるMCEaの年換算発現率(平均値)は、ドジョルビ投与前bが1.76回/年、ドジョルビ投与開始後cが1.00回/年で、減少率は43.5%であった(p=0.0347、対応のあるt検定、名目上のp値)。
副次評価項目であるMCEaの年換算発現日数(中央値d)は、ドジョルビ投与前bが5.33日/年、ドジョルビ投与開始後cが2.93日/年で、減少率は45.1%であった(p=0.8139、Wilcoxon符号付順位検定、名目上のp値)。
トリヘプタノインナイーブコホートにおけるMCEの年換算発現率及び年換算発現日数(FAS)〈主要評価項目及び副次評価項目〉
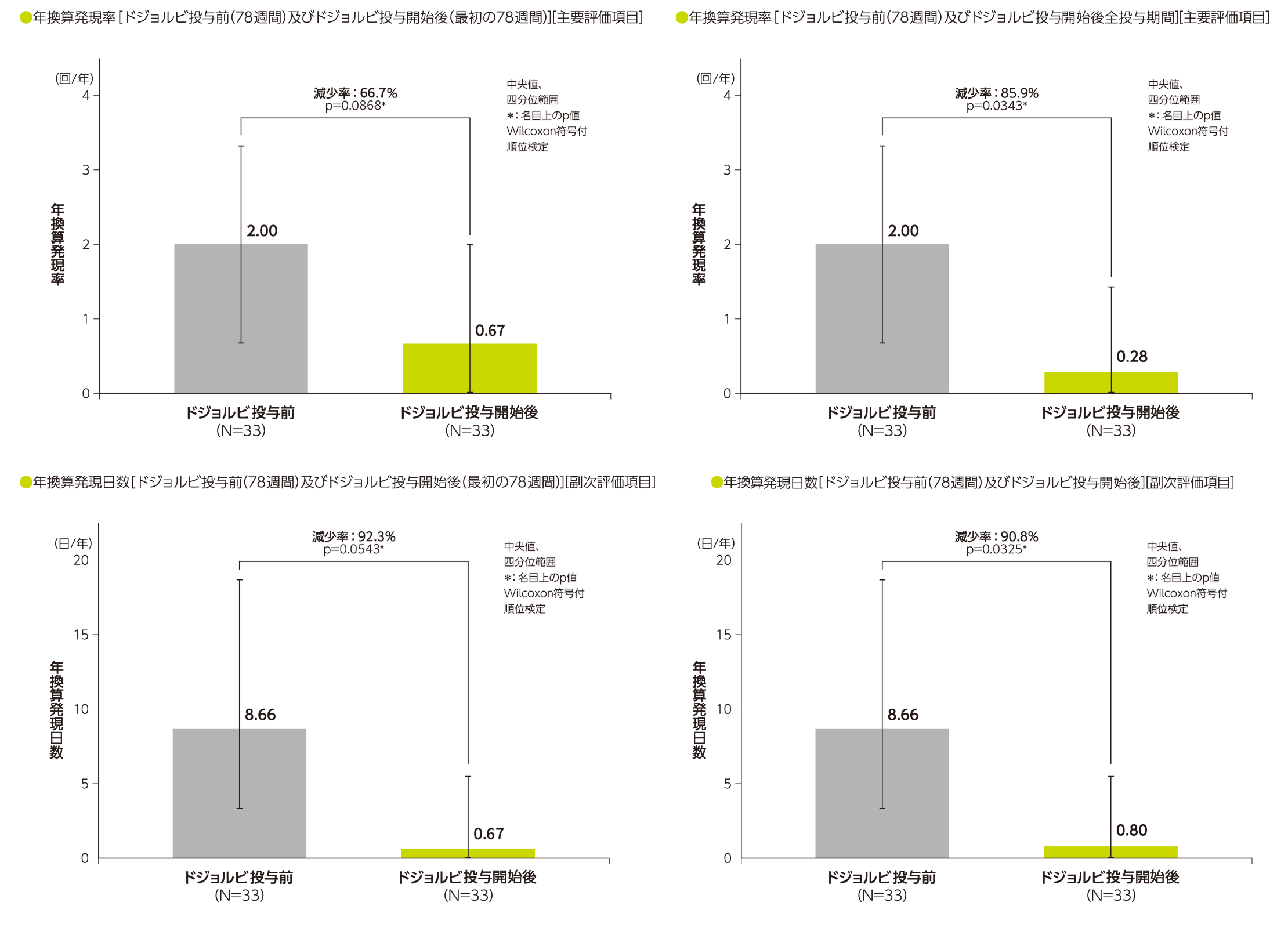
トリヘプタノインナイーブコホートeにおいて、主要評価項目であるMCEaの年換算発現率f(中央値d)は、ドジョルビ投与前が2.00回/年、ドジョルビ投与開始後最初の78週間gが0.67回/年、投与開始後全投与期間が0.28回/年で、減少率はそれぞれ66.7%(p=0.0868、Wilcoxon符号付順位検定、名目上のp値)、85.9%(p=0.0343、Wilcoxon符号付順位検定)であった。
副次評価項目である年換算発現日数h(中央値d)は、ドジョルビ投与前が8.66日/年、ドジョルビ投与開始後最初の78週間gが0.67日/年、投与開始後全期間が0.80日/年で、減少率はそれぞれ92.3%(p=0.0543、Wilcoxon符号付順位検定、名目上のp値)、90.8%(p=0.0325、Wilcoxon符号付順位検定、名目上のp値)であった。
各MCEの年換算発現率および発現日数
UX007-CL201試験移行コホートにおける各MCEの年換算発現率(FAS)[ドジョルビ投与前(78週間)及びドジョルビ投与開始後]〈副次評価項目〉
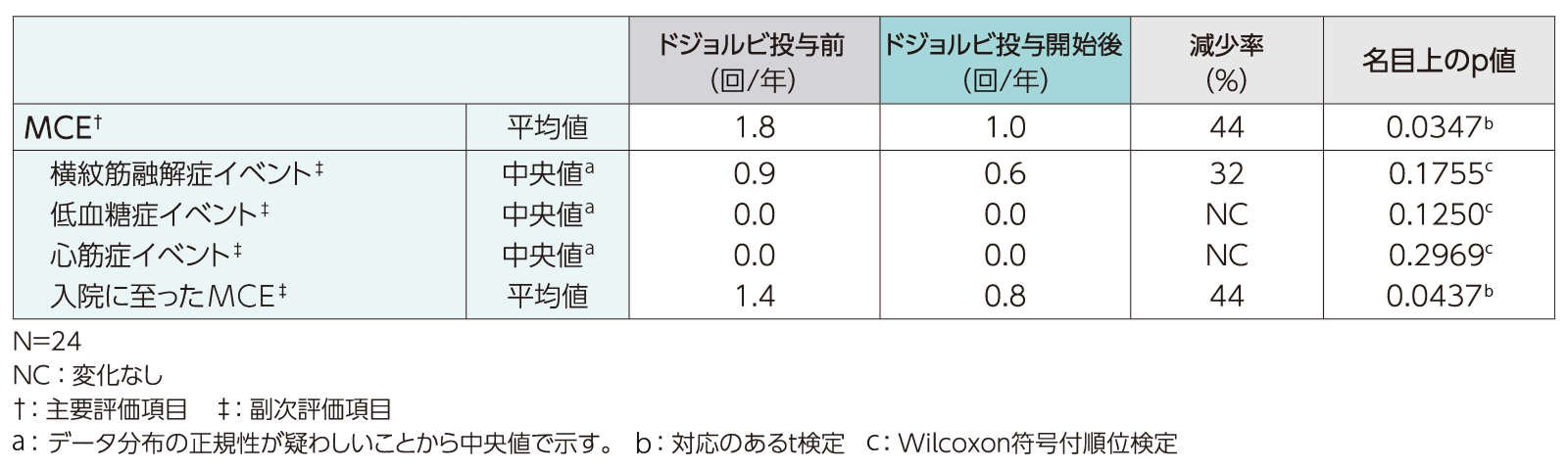
UX007-CL201試験移行コホートにおける各MCEの年換算発現率は、横紋筋融解症イベントはドジョルビ投与前dが0.9回/年、ドジョルビ投与開始後eが0.6回/年(各中央値)で、減少率は32%であった(p=0.1755、Wilcoxon符号付順位検定、名目上のp値)。入院に至ったMCEfは、ドジョルビ投与前が1.4回/年、ドジョルビ投与開始後eが0.8回/年(各平均値)で減少率は44%であった。(p=0.0437、対応のあるt検定、名目上のp値)低血糖症イベント及び心筋症イベントの年換算発現率(中央値)は、上表の通りであったg。
UX007-CL201試験移行コホートにおける各MCEの年換算発現日数(FAS)[ドジョルビ投与前(78週間)及びドジョルビ投与開始後]〈副次評価項目〉
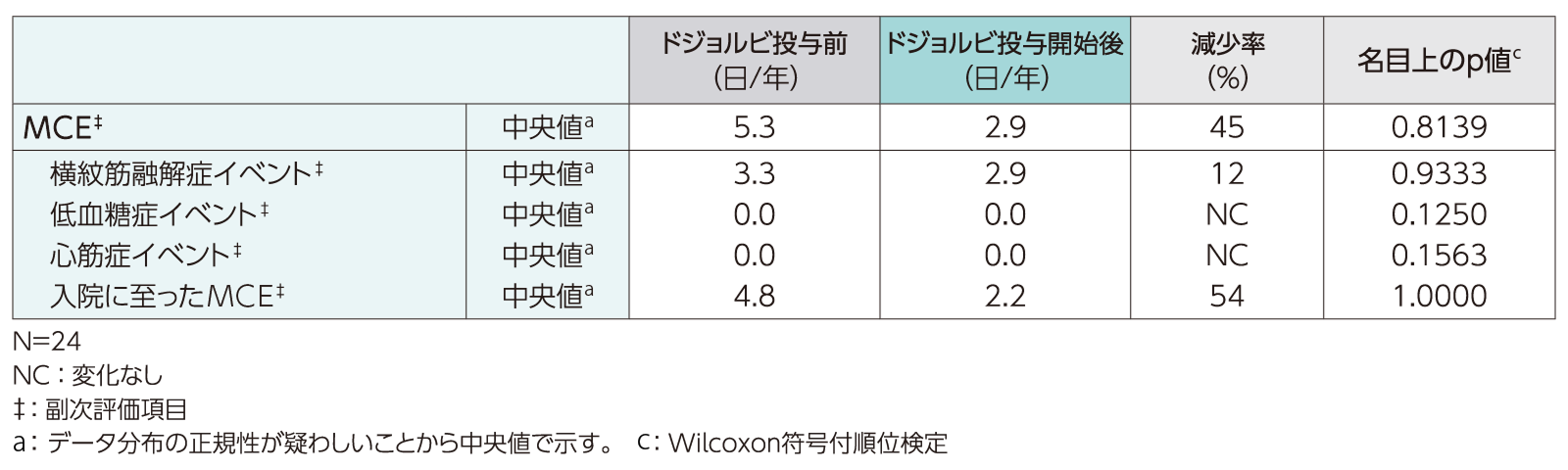
UX007-CL201試験移行コホートにおける各MCEの年換算発現日数は、横紋筋融解症イベントはドジョルビ投与前dが3.3日/年、ドジョルビ投与開始後eが2.9日/年(各中央値)で、減少率は12%であった(p=0.9333、Wilcoxon符号付順位検定、名目上のp値)。入院に至ったMCEfは、ドジョルビ投与前が4.8日/年、ドジョルビ投与開始後eが2.2日/年(各中央値)で、減少率は54%であった(p=1.0000、Wilcoxon符号付順位検定、名目上のp値)。低血糖症イベント及び心筋症イベントの年換算発現日数(中央値)は上表のとおりであったc。
トリヘプタノインナイーブコホートにおける各MCEの年換算発現率(FAS)[ドジョルビ投与前(78週間)及びドジョルビ投与開始後(全投与期間)]〈副次評価項目〉
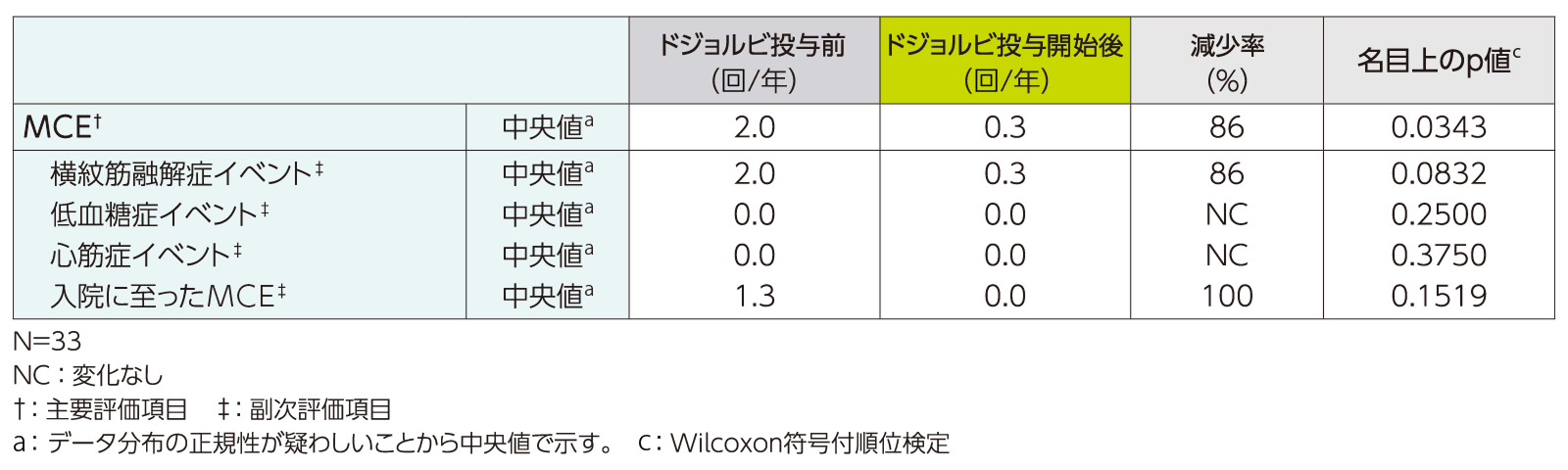
トリヘプタノインナイーブコホートhにおける各MCEの年換算発現率は、横紋筋融解症イベントは、ドジョルビ投与前が2.0回/年、ドジョルビ投与開始後iが0.3回/年(各中央値)で、減少率は86%であった(p=0.0832、Wilcoxon符号付順位検定、名目上のp値)。入院に至ったMCEfは、ドジョルビ投与前が1.3回/年、ドジョルビ投与開始iが0.0回/年(各中央値)で、減少率は100%であった(p=0.1519、Wilcoxon符号付順位検定、名目上のp値)。低血糖症イベント及び心筋症イベントの年換算発現率(中央値)は上表のとおりであったe。
トリヘプタノインナイーブコホートにおける各MCEの年換算発現日数(FAS)[ドジョルビ投与前(78週間)及びドジョルビ投与開始後(全投与期間)]〈副次評価項目〉
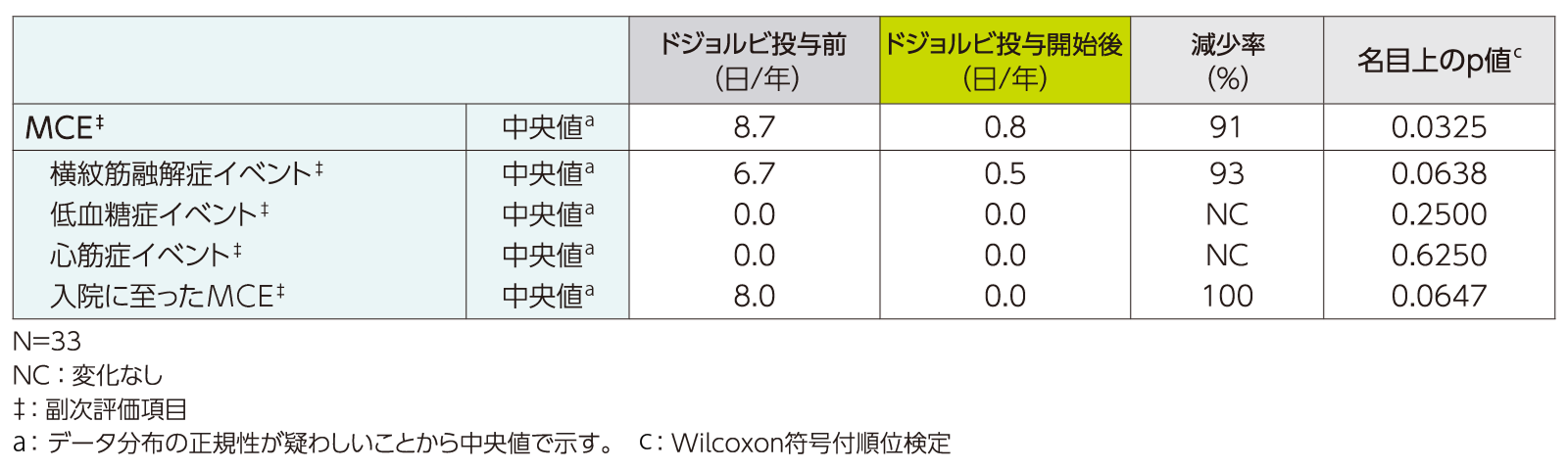
トリヘプタノインナイーブコホートhにおける各MCEの年換算発現日数は、横紋筋融解症イベントは、ドジョルビ投与前が6.7日/年、ドジョルビ投与開始後iが0.5日/年(各中央値)で、減少率は93%であった(p=0.0638、Wilcoxon符号付順位検定、名目上のp値)。入院に至ったMCEfは、ドジョルビ投与前が8.0日/年、ドジョルビ投与開始後iが0.0日/年(各中央値)で、減少率は100%であった(p=0.0647、Wilcoxon符号付順位検定、名目上のp値)。低血糖症イベント及び心筋症イベントの年換算発現日数(各中央値)は上表の通りであったi。
副作用
副作用(全体で2例以上の患者に発現した副作用)(安全性解析対象集団)
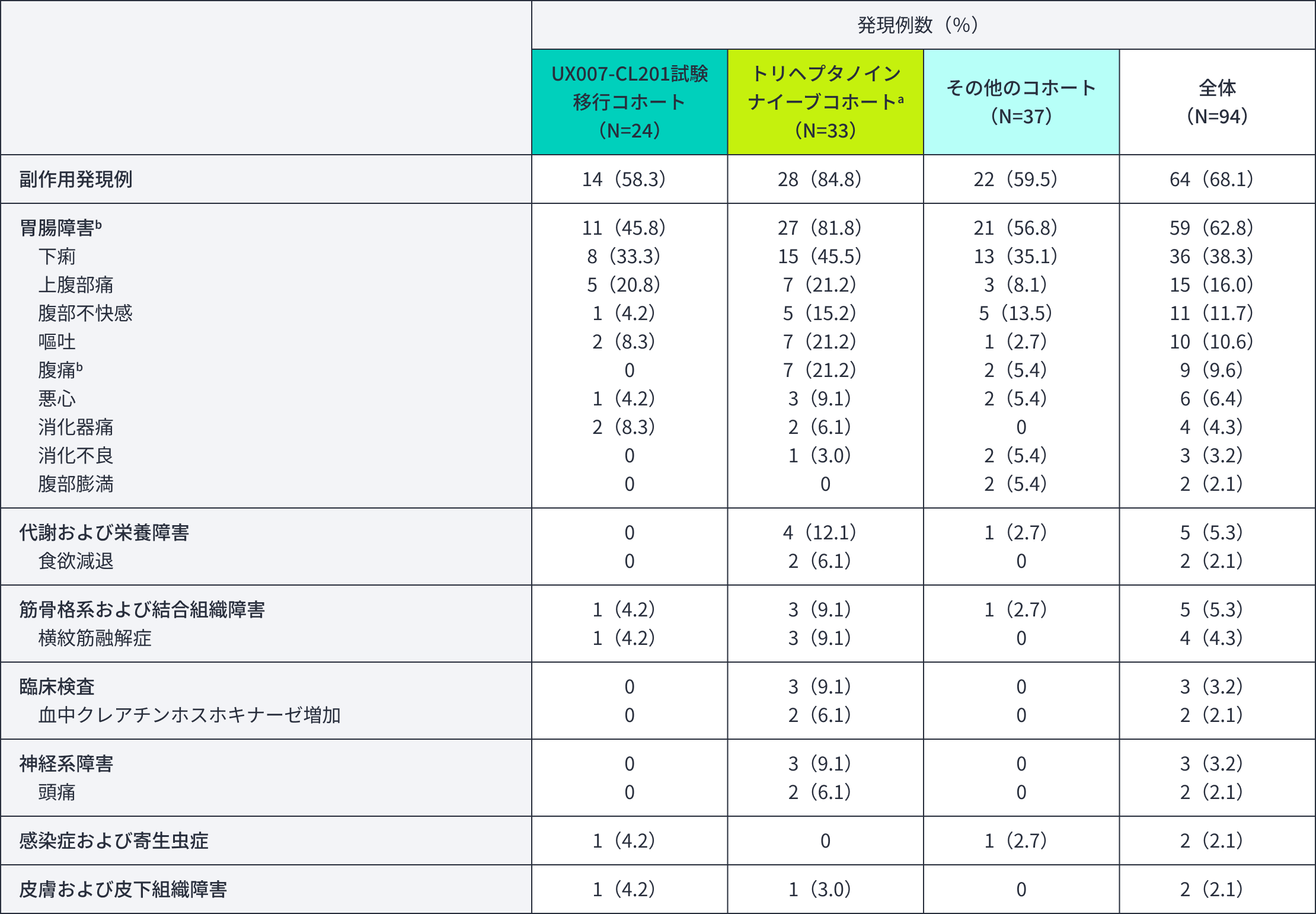
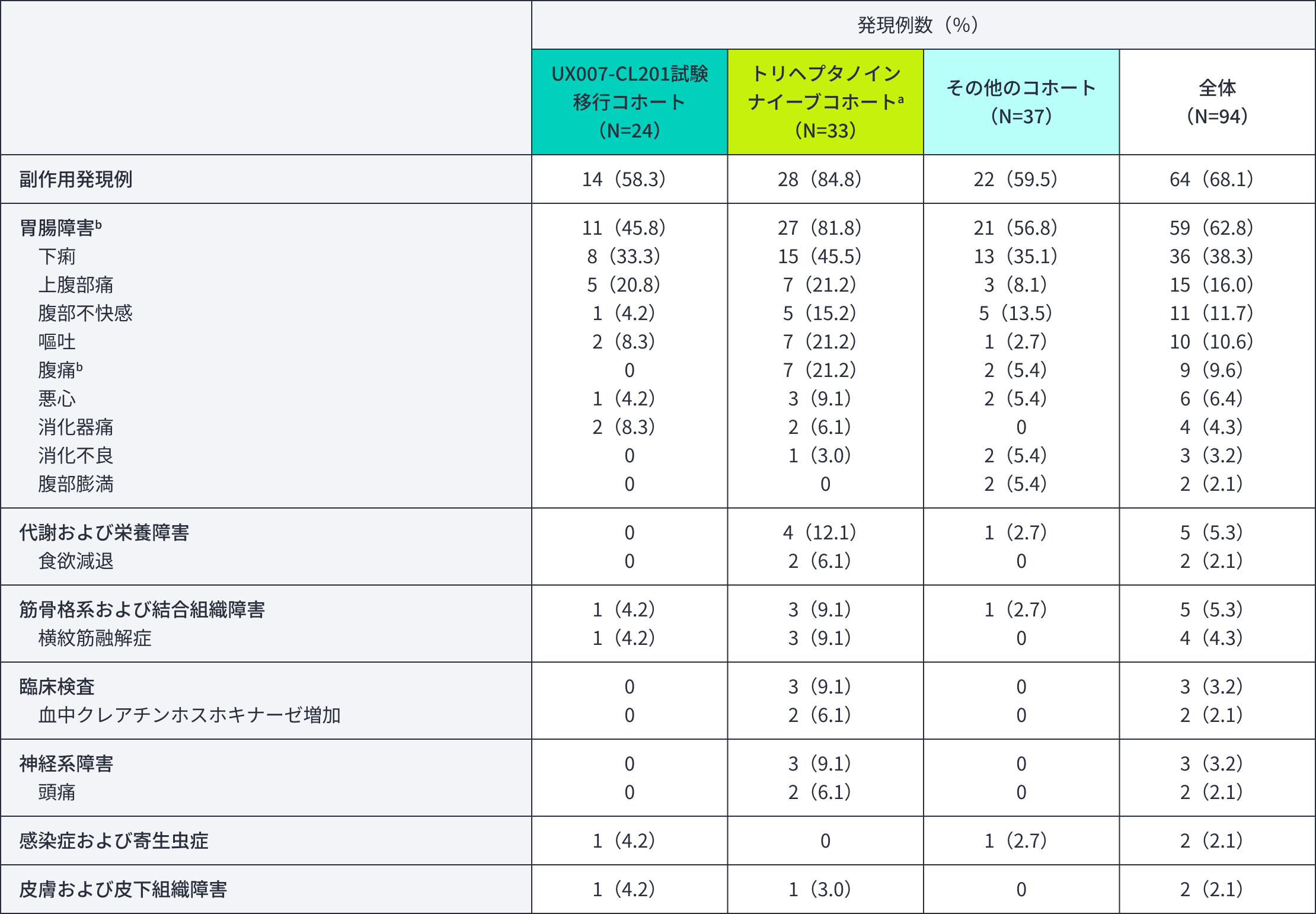
| 発現例数(%) | ||||
|---|---|---|---|---|
| UX007-CL201 試験移行コホート (N=24) | トリヘプタノイン ディープコホート (N=33) | その他のコホート (N=37) | 全体 (N=94) | |
| 副作用発現例 | 14 (58.3) | 28 (84.8) | 22 (59.5) | 64 (68.1) |
| 胃腸障害* | ||||
| 下痢 | 11 (45.8) | 27 (81.8) | 21 (56.8) | 59 (62.8) |
| 嘔吐 | 8 (33.3) | 15 (45.5) | 13 (35.1) | 36 (38.3) |
| 上腹部痛 | 5 (20.8) | 7 (21.2) | 3 (8.1) | 15 (16.0) |
| 腹部不快感 | 1 (4.2) | 5 (15.2) | 5 (13.5) | 11 (11.7) |
| 嘔気 | 2 (8.3) | 7 (21.2) | 1 (2.7) | 10 (10.6) |
| 腹痛* | 0 | 7 (21.2) | 2 (5.4) | 9 (9.6) |
| 悪心 | 1 (4.2) | 3 (9.1) | 2 (5.4) | 6 (6.4) |
| 消化不良 | 2 (8.3) | 2 (6.1) | 0 | 4 (4.3) |
| 腹部膨満 | 0 | 1 (3.0) | 2 (5.4) | 3 (3.2) |
| 腹部膨満感 | 0 | 0 | 2 (5.4) | 2 (2.1) |
| 代謝および栄養障害 | ||||
| 食欲減退 | 0 | 4 (12.1) | 1 (2.7) | 5 (5.3) |
| 脱水 | 0 | 2 (6.1) | 0 | 2 (2.1) |
| 筋骨格系および結合組織障害 | ||||
| 筋肉痛 | 1 (4.2) | 3 (9.1) | 1 (2.7) | 5 (5.3) |
| 筋痙縮 | 1 (4.2) | 3 (9.1) | 0 | 4 (4.3) |
| 臨床検査 | ||||
| 血中クレアチンホスホキナーゼ増加 | 0 | 3 (9.1) | 0 | 3 (3.2) |
| 体重減少 | 0 | 2 (6.1) | 0 | 2 (2.1) |
| 神経系障害 | ||||
| 頭痛 | 0 | 3 (9.1) | 0 | 3 (3.2) |
| 浮動性めまい | 0 | 2 (6.1) | 0 | 2 (2.1) |
| 感染症および寄生虫症 | 1 (4.2) | 0 | 1 (2.7) | 2 (2.1) |
| 皮膚および皮下組織障害 | 1 (4.2) | 1 (3.0) | 0 | 2 (2.1) |
MedDRA version 20.0 器官別大分類(SOC)/PT
副作用cが1件以上発現した患者は、94例中64例(68.1%)であった(安全性解析対象集団)。主な副作用(発現率5%以上)は、下痢が36例(38.3%)、上腹部痛が15例(16.0%)、腹部不快感が11例(11.7%)、嘔吐が10例(10.6%)、腹痛が9例(9.6%)、悪心が6例(6.4%)であった。重篤な副作用はUX007-CL201試験移行コホートで4.2%(1/24例、イレウス)、トリヘプタノインナイーブコホートで9.1%[3/33例、横紋筋融解症及び急性膵炎各1例、横紋筋融解症・慢性胃炎・胃食道逆流性疾患1例(同一症例)]、その他のコホートで2.7%(1/37例、憩室炎)に認められた。治験薬の投与中止に至った副作用の発現はその他のコホート1例に認められ、筋力低下であった。
本試験では、UX007-CL201試験移行コホートで8.3%(2/24例、心不全及び心肺停止各1例)、トリヘプタノインナイーブコホートで3.0%(1/33例、心肺停止)、その他のコホートで5.4%(2/37例、心不全及び心筋症の悪化各1例)の死亡が報告された。副作用による死亡例はなかった。
目的
LC-FAOD(すべてのサブタイプ)患者におけるドジョルビの長期安全性及び有効性を評価する。
試験デザイン
後ろ向き及び前向き、介入、非盲検、単群、被験者自己対照試験
対象
LC-FAOD(すべてのサブタイプ)患者94例
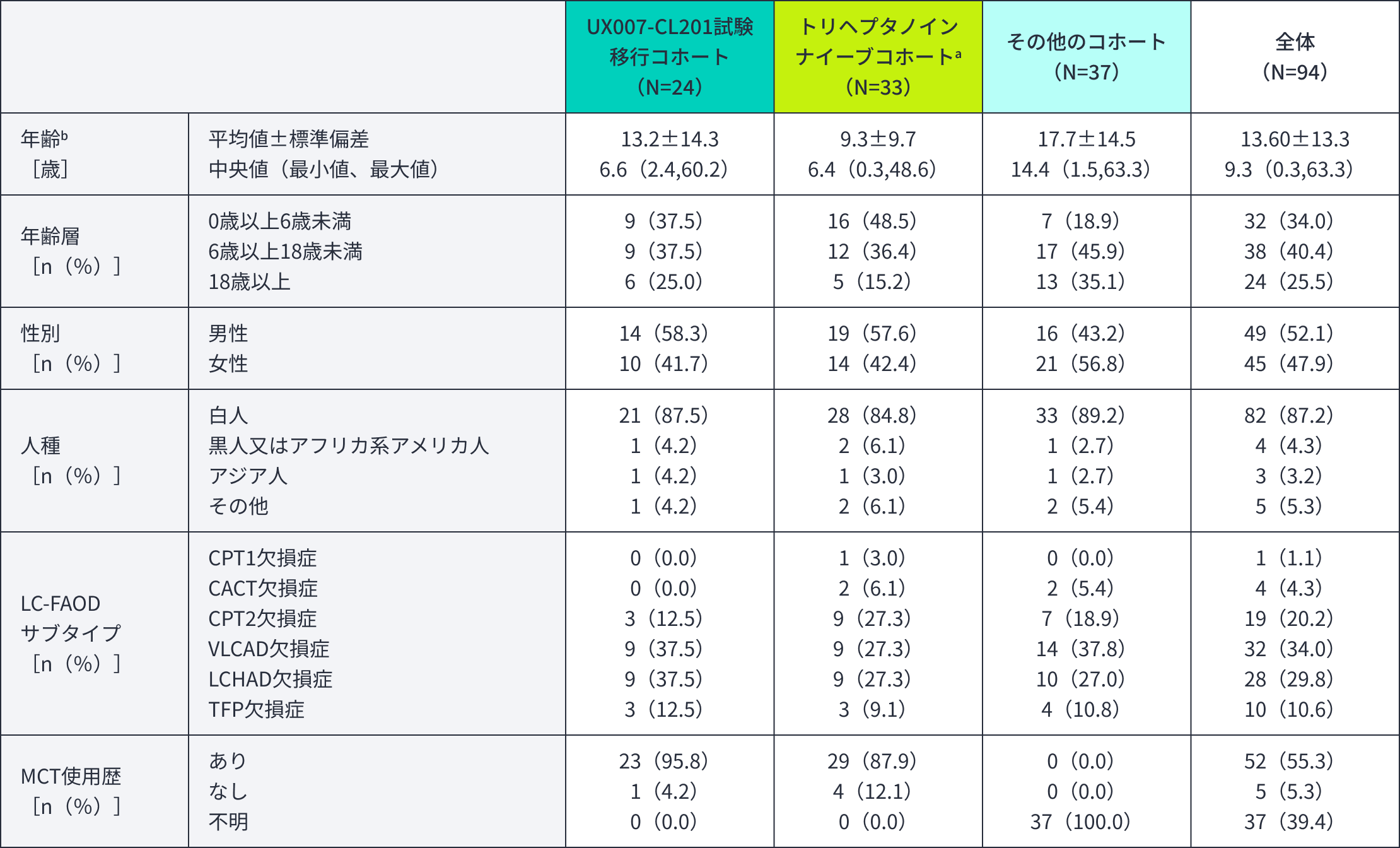
- UX007-CL201試験から移行した患者(UX007-CL201試験移行コホート)24例
- トリヘプタノインの投与歴がない患者(トリヘプタノインナイーブコホート)33例a
- 医師主導治験(IST)又はその他の治療プログラムから移行した患者(その他のコホート)37例
〈主な選択基準〉
- CPT1若しくはCPT2欠損症、VLCAD欠損症、LCHAD欠損症、TFP欠損症又はCACT欠損症のいずれかと確定診断された者
- 生後6ヵ月以上の者
- UX007-CL201試験を完了した者(UX007-CL201試験移行コホート)
- トリヘプタノイン未投与で、従来の治療が無効であり、重度のアンメットニーズが確認されている者(トリヘプタノインナイーブコホート)
- IST又は他の方法を通じてトリヘプタノインを投与された者(その他のコホート)
〈主な除外基準〉
- MCAD欠損症、短鎖又は中鎖FAOD、ケトン体代謝異常症、プロピオン酸血症若しくはメチルマロン酸血症と診断された者
- LC-FAODにおけるトリヘプタノインの別の臨床試験に適格となった者
方法
ドジョルビの投与量は、1日あたりのカロリー摂取量(DCI)aの25~35%を目標とし、1日4回(朝食、昼食、夕食、就寝前)に分け、食物又は飲料(乳児用粉ミルクを含む)に混合して経口又は経管投与した。開始用量はDCIの約10%とし、目標投与量に至るまで忍容性を確認しながら漸増した。また、ベースライン時にMCTを使用していた患者では、MCTの量と同量からの開始を可能とし、ベースライン時にMCTの投与を中止した。
ドジョルビへの忍容性が認められない場合は、1日あたりの投与回数を増加又は経管投与時の投与時間を延長し、それでも忍容性が認められない場合には減量が可能とされ、その後は最大耐用量を維持した。本試験では、以下の増量及び減量に係る規定が推奨されていた。また、各被験者の状況に応じて柔軟な対応も認められていた。
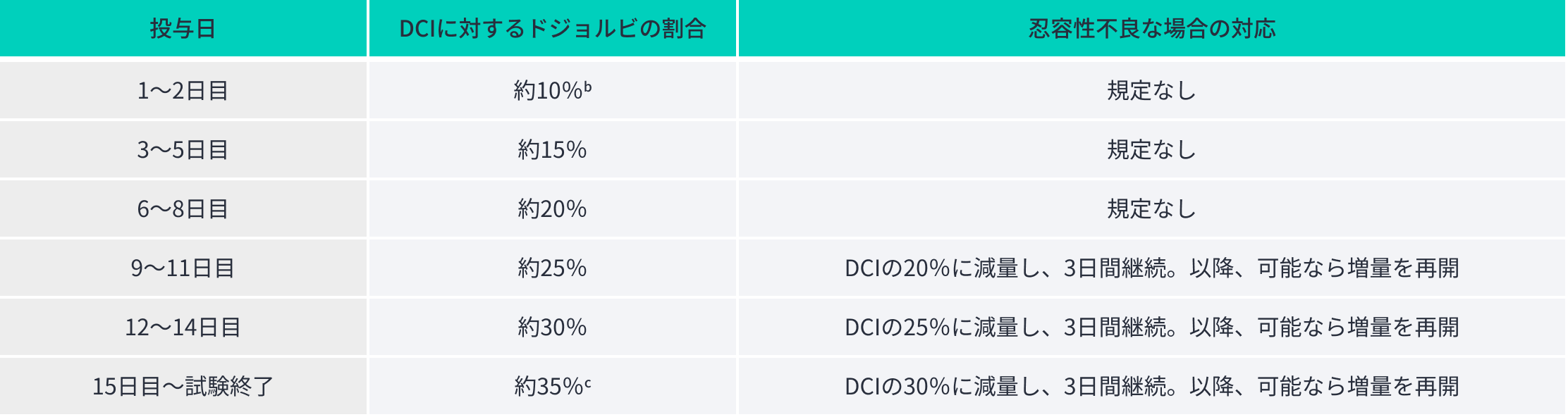
投与期間は約7年(84ヵ月)又はドジョルビ承認のいずれか早い時点までとした。
最終投与の30~35日後に安全性追跡調査を実施した。UX007-CL201試験移行コホートのベースラインは、UX007-CL201試験の最終来院時(78週時)に実施した評価を使用した。
本試験では、UX007-CL201試験移行コホート及びトリヘプタノインナイーブコホートを対象として、ドジョルビ投与前78週間及びドジョルビ投与開始後のMCEd(定義については脚注d参照)発現について、患者内比較を行った。なお、その他のコホートは、過去18~24ヵ月間のMCEdデータは収集せず、本試験での長期安全性を評価した。
UX007-CL202試験のデザイン
食事日誌に基づき、1日あたりの平均カロリー摂取量を算出した。
評価項目
〈有効性評価項目〉
主要評価項目:
- UX007-CL201試験移行コホートにおけるドジョルビ投与前78週間a及びドジョルビ投与開始後bのMCEcの年換算発現率d
- トリヘプタノインナイーブコホートにおけるドジョルビ投与前78週間a及びドジョルビ投与開始後のMCEcの年換算発現率d(全投与期間及び78週間) 等
副次評価項目:
- UX007-CL201試験移行コホートにおけるドジョルビ投与前78週間a及びドジョルビ投与開始後bのMCEcの年換算発現日数e
- トリヘプタノインナイーブコホートにおけるドジョルビ投与前78週間a及びドジョルビ投与開始後のMCEcの年換算発現日数e(全投与期間及び78週間)
- UX007-CL201試験移行コホートにおけるドジョルビ投与前78週間a及びドジョルビ投与開始後の各MCEfの年換算発現率d及び年換算発現日数e
- 横紋筋融解症イベント、低血糖症イベント、心筋症イベント、入院に至ったMCE
- トリヘプタノインナイーブコホートにおけるドジョルビ投与前78週間a及びドジョルビ投与開始後gの各MCEfの年換算発現率d及び年換算発現日数e
- 横紋筋融解症イベント、低血糖症イベント、心筋症イベント、入院に至ったMCE 等
〈安全性評価項目〉
- 副作用 等
解析計画
〈解析対象集団〉
| 定義 | 例数 | |
|---|---|---|
| 最大の解析 対象集団(FAS) |
ドジョルビ投与後、少なくとも1回有効性の評価が実施されたす べての患者 |
UX007-CL201試験 移行コホート:24 トリヘプタノイン ナイーブコホート:33 |
| 安全性解析対象集団 | 本試験中にドジョルビを少なくとも1回投与されたすべての患者 | 94 |
〈有効性評価項目〉
有効性データの解析には、FASを用いた。UX007-CL201試験移行コホートは、UX007-CL201試験期間及びUX007-CL202試験期間で収集したデータを併合した。
主要評価項目は、データの分布に応じて平均値又は中央値で示し、ドジョルビ投与前及びドジョルビ投与開始後を、それぞれ対応のあるt検定又はWilcoxon符号付順位検定を用いて比較した。
副次評価項目は、主要評価項目と同様の手法で解析した。
検定は有意水準を両側5%とし、多重性の調整は実施しなかった。
〈安全性評価項目〉
安全性データの解析には、安全性解析対象集団を用いた。安全性データは記述的に要約した。副作用名はMedDRA version 20.0でコーディングした。
患者背景
患者背景(安全性解析対象集団)
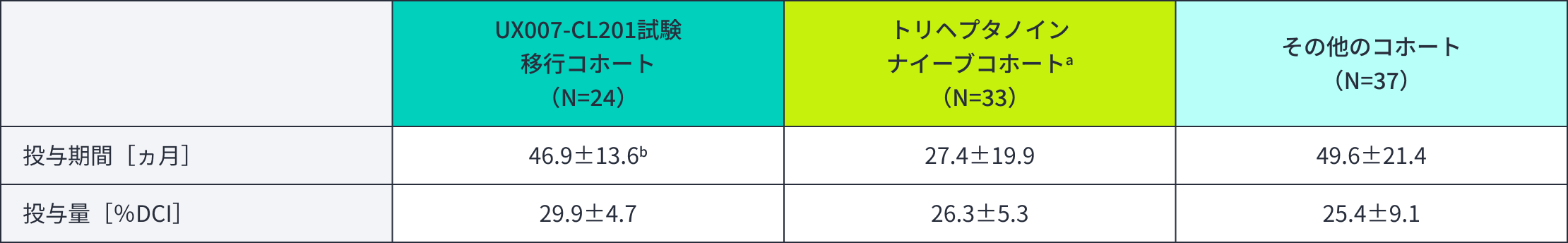
曝露及び投与量
曝露及び投与量(安全性解析対象集団)
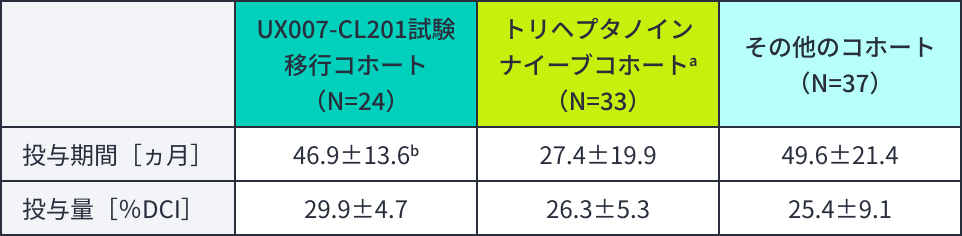
平均値±標準偏差
参考文献
- 社内資料:UX007-CL202試験/海外第Ⅱ相試験(CTD2.7.3.2.2、2.7.4.2.1、2.7.6.2.3、5.3.5.2)[承認時評価資料]
- Vockley J, et al.: J Inherit Metab Dis. 2021; 44(1):253-263.(利益相反:Ultragenyx Pharmaceutical, Incの支援により実施)
- Vockley J, et al.: J Inherit Metab Dis. 2023; 46(5):943-955.(利益相反:Ultragenyx Pharmaceutical, Incの支援により実施)